
Lessons from our Pupils: A Reflection [Podcast Episode 147]
In January 2nd’s Episode (LINK), Jay was joined by Dr. Allen Ho to discuss his recently published paper in Nature Medicine regarding the successful use of an intravitreal oligonucleotide for a form of Leber Congenital Amaurosis (LCA). Dr. Matthew Weed then joined the podcast to discuss the article and compare previously used therapies like Luxturna. In this post we are going to review what Leber Congenital Amaurosis is and how this new oligonucleotide functions to rescue one of the mutations that causes LCA.
Leber Congenital Amaurosis is a genetic disorder that primarily affects the retina leading to visual impairment beginning in infancy. Patients suffering from this disorder can also have nystagmus, photophobia, and slow pupillary reactions. Visual impairment begins in early childhood, and progressively deteriorates ultimately resulting in vision loss at around thirty to forty years old. This disorder is inherited primarily in an autosomal recessive pattern. Mutations in at least 14 genes have been identified, with the most common being CEP290, CRB1, GUCY2D, and RPE65. You may have read or listened to our recent discussion on the use of gene therapy for RPE65 (LINK TO PODCAST AND BLOG). The article discussed in Episode 147 focused on CEP290, which is a gene that plays an important role in the development of centrosomes and cilia. The most common mutation in CEP290 that leads to LCA causes a splicing error in pre-mRNA. The mutation, which changes an adenine to a guanine, occurs within one of the introns of CEP290. This creates a new splice-donor site and a new exon (Exon 10) is aberrantly inserted into the final mRNA. This new exon carries a premature stop codon that, when translated, results in a truncated CEP290 protein that no longer functions as the original protein.
An antisense oligonucleotide (ASO) is a short (generally 13-25 nucleotides) single-stranded DNA molecule that can hybridize to a unique target sequence in a cell. The first generation of ASOs were designed to target mRNA and thereby knockdown the transcript via endonuclease-mediated degradation. These agents had the disadvantage of fast turnover, which prohibited them from achieving intracellular concentrations sufficient to suppress their target. New ASOs have been developed with modified backbones that function through different mechanisms like preventing ribosome recruiting to inhibit translation, or sterically blocking splicing factors to alter pre-mRNA splicing.

The ASO (QR-110) used in the paper discussed in Episode 147 works by modifying splicing in a slightly different manner than described above. QR-110 binds to the CEP290 pre-mRNA at the intron that contains the mutation. This binding prevents the creation of a new splice site, which causes the pre-mRNA to be processed as the wild-type pre-mRNA, without the inclusion of Exon 10. The protein translated from the mRNA is a wild-type CEP290 protein that can function normally in the development of centrosomes and cilia.
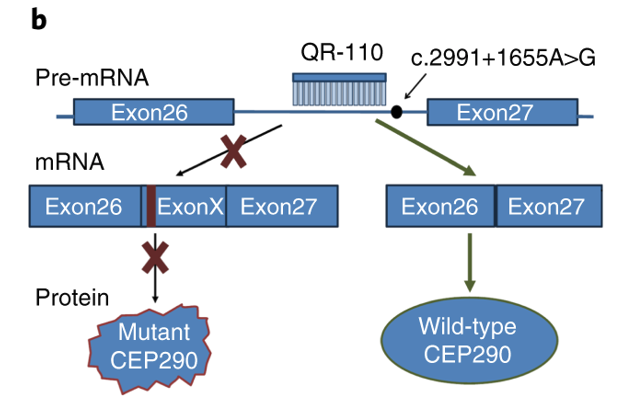
Image credit: https://www.nature.com/articles/s41591-018-0295-0#Fig1
-Amy Kloosterboer